Naming Pseudogenes
HGNC ·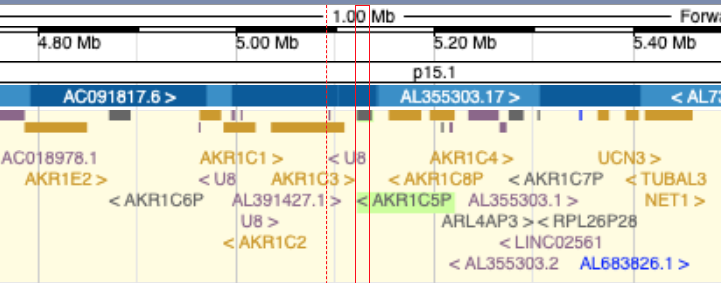
In amongst the functional genes encoding proteins, the human genome contains a myriad of non-functional gene copies and gene fragments – known as pseudogenes. By naming these pseudogenes we help to reveal their relationships to their functional gene relatives, and to ‘label’ the genome more fully . However, naming every bit of genetic shrapnel would be a Herculean and arguably pointless task, so how do we decide which pseudogenes to name - and how do we name them?
Pseudogene origins
Before we consider these questions, it is worth reminding ourselves of the possible routes that generate pseudogenes:
- Reverse transcription of mRNA from a parent gene followed by integration into the genome – the resulting ‘processed’ pseudogenes are characterized by having no introns and are often incomplete at the 5’ end.
- Duplication of the parent followed by mutation, or partial duplication – duplicated pseudogenes retain introns and are often found beside functional copies.
- Mutation – a non-essential single copy gene may lose the ability to encode a protein due to mutation. These pseudogenes, which have no ‘parent’ gene in the genome, are sometimes called ‘unitary’ pseudogenes. Note that the equivalent gene may be functional in other species.
Don’t sweat the small stuff
In general, we do not name pseudogenes that are very degraded, i.e. those that retain only a very small proportion of the original coding sequence, or where the identity of the parent gene is unclear. However, we do not apply strict percentage cut-offs – each sequence is considered on a case-by-case basis, e.g. a publication on the pseudogene or a researcher request for a name could trump our qualms about the length or percentage identity.
Transcribed v non-transcribed
We name both transcribed and non-transcribed pseudogenes, but prioritize those that are transcribed on the grounds they are more likely to do something.
How do we name pseudogenes?
You may have noticed that there are different styles of pseudogene names – they may be named as a numbered pseudogene of a particular parent gene, e.g. OTX2P2, or given a separate number within a series of related genes, e.g. AKR1C5P. The pseudogenes named within a series typically represent pseudogenes that are almost intact and may have originally been protein coding. Historically, separate numbers have also been used for each member of a duplicated gene array even if some members of the array are short and/or degraded.
“I’ll have a ‘P’ please Bob” §
In both naming styles, we include the word ‘pseudogene’ in the gene name and usually a ‘P’ for pseudogene in the gene symbol (we may make a rare exception when the pseudogene has been published using a symbol that does not contain P). However, P is not reserved exclusively for ‘pseudogene’ - so don’t assume that every gene symbol ending in P, or P followed by a number, is a pseudogene. [Check the locus type to be certain!]
To a large extent the different styles reflect the origin of the pseudogene – the majority of processed pseudogenes take the symbol of the parent gene followed by P1, P2 etc (e.g. Figure 1 GENE1P1) whereas duplicated and unitary pseudogenes tend to be given a separate number followed by P (e.g. Figure 1 GENE3P). However, there are some exceptions: for example, if a processed pseudogene looks like it has the potential to be a functional “retrogene”, i.e. a gene copy duplicated via retrotransposition, it would be given a separate number in a series, then making it easy for us simply to drop the ‘P’ if it later does prove to be functional (e.g. Figure 1 GENE2P). Also, very short or substantially degraded pseudogenes without orthologs in other species are named as a numbered pseudogene of their closest homolog in the genome, regardless of whether they are processed or unprocessed (e.g. Figure 1 GENE1P2).
How are pseudogene symbols related between species?
It is not unusual for duplicated pseudogenes that appear in clusters with active gene family members to be conserved across species but sometimes with different copies functional in different species. Where possible orthologous copies are given the same symbol with P appended to the pseudogenized genes, but this is not always the case.
In contrast, there is no significance to the numbers in the P1, P2 style named pseudogene series – the numbers simply reflect the order of naming. No orthologous relationships can be inferred from the numbering of pseudogenes in other species as this numbering is usually species-specific.
Where do our pseudogenes come from?
Many of our current pseudogenes were named based on data from pseudogene.org, whose data is now integrated into the GENCODE gene set which is displayed in Ensembl. Hence we review unnamed pseudogenes in Ensembl and compare these with NCBI Gene annotations before assigning names. Generally, these annotation groups provide a clue to the parent gene – but we also do our own sequence searches to check what we think the closest functional relatives are. In this process we would usually ignore short matches (< 20% coverage of the parent gene) but would name any new pseudogenes that are longer than those already named for that gene. Sometimes there is no clear parent of a processed pseudogene – in these cases we may choose to name it based on a root symbol rather than a specific gene, e.g. ACTP1 is equally homologous to several actin genes.
We also name pseudogenes that we spot in publications and based on requests from curators at other databases. We rarely receive requests from researchers to name pseudogenes…but please feel free to ask!
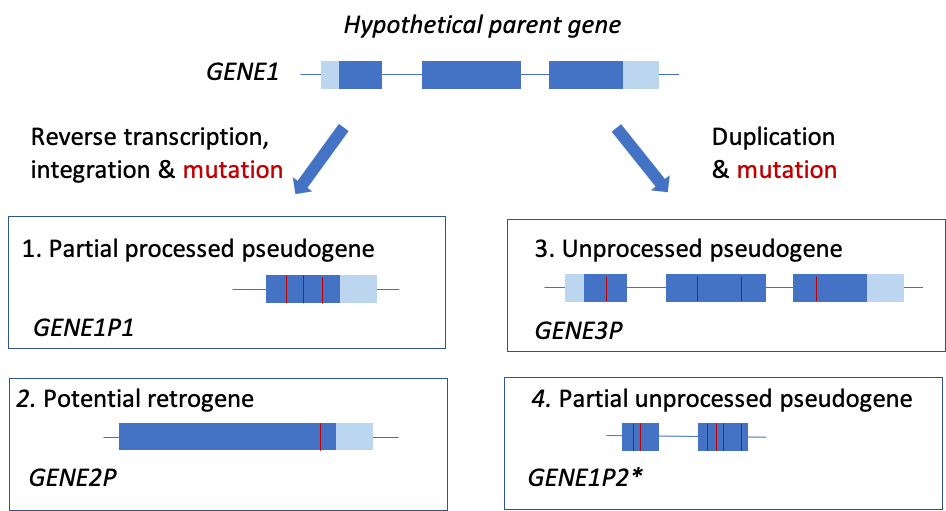
This figure illustrates how we would name 4 possible pseudogenes derived from the hypothetical parent gene GENE1. *If this gene was in a cluster with GENE1 and GENE3P, we’d call it GENE4P. Dark blue boxes represent coding regions in the parent gene and pale blue boxes untranslated regions. Red lines represent mutations.
§ A reference to the UK quiz show Blockbusters hosted by Bob Holness.