Transcripts Are The Mane Attraction
HGNC, Guest Post ·This guest blog post has been contributed by Jane Loveland who works in the Ensembl-Havana gene annotation group who are long term collaborators of the HGNC.

I’m an Annotation Project Leader in the Ensembl-Havana team at EMBL-EBI and have been with the team for almost 20 years (!). We manually annotate genes in the human and mouse genomes as part of the GENCODE project.
My first job in science was in plant virus research, which I applied for in a panic on my return after a gap year in Australia following my degree (in biology and geography), when I realized I was back in the real world. I surprised myself by really enjoying it and followed it up with a move to a biochemistry group, where I undertook a PhD in plant biochemistry and molecular biology. I then left wet lab work to work in bioinformatics as a computational biology specialist for the BBSRC, visiting lab groups and delivering training at 8 BBSRC sites.
My next move was to the Havana team, initially based at the Wellcome Sanger Institute. I started in 2002 when the first draft of the human genome had only recently been released, and we were annotating genes on the newly sequenced chromosomes. This work resulted in the high-profile Nature chromosome publications, and I found it exciting and a bit daunting after coming from a quiet plant lab! Many of these genes we were annotating didn’t have symbols or names, and so our collaboration with HGNC began.
Automated gene annotation from Ensembl is a good first stage in the analysis of genomes, but for high value reference species, such as human and mouse, accuracy is essential and so manual gene annotation is required. We annotate by aligning sets of transcripts to the genome to find the best-in-genome location. These data can be cDNAs and ESTs, or more likely these days long read transcriptomic data (PacBio, SLR-Seq, ONT). We then assign each locus we annotate a biotype, such as protein-coding, long non-coding RNA (lncRNA), pseudogene and anything in-between, but to a defined set of guidelines. We call them guidelines rather than rules, like they do for gene naming, as in biology we often find things that break any rules! Once we’ve checked and added the symbol and name at HGNC, we then release our GENCODE geneset through genecodegenes.org. This is the default geneset for human and mouse in the Ensembl browser and the UCSC genome browser. If we discover a new gene or find an unnamed gene, we contact HGNC to start a discussion.
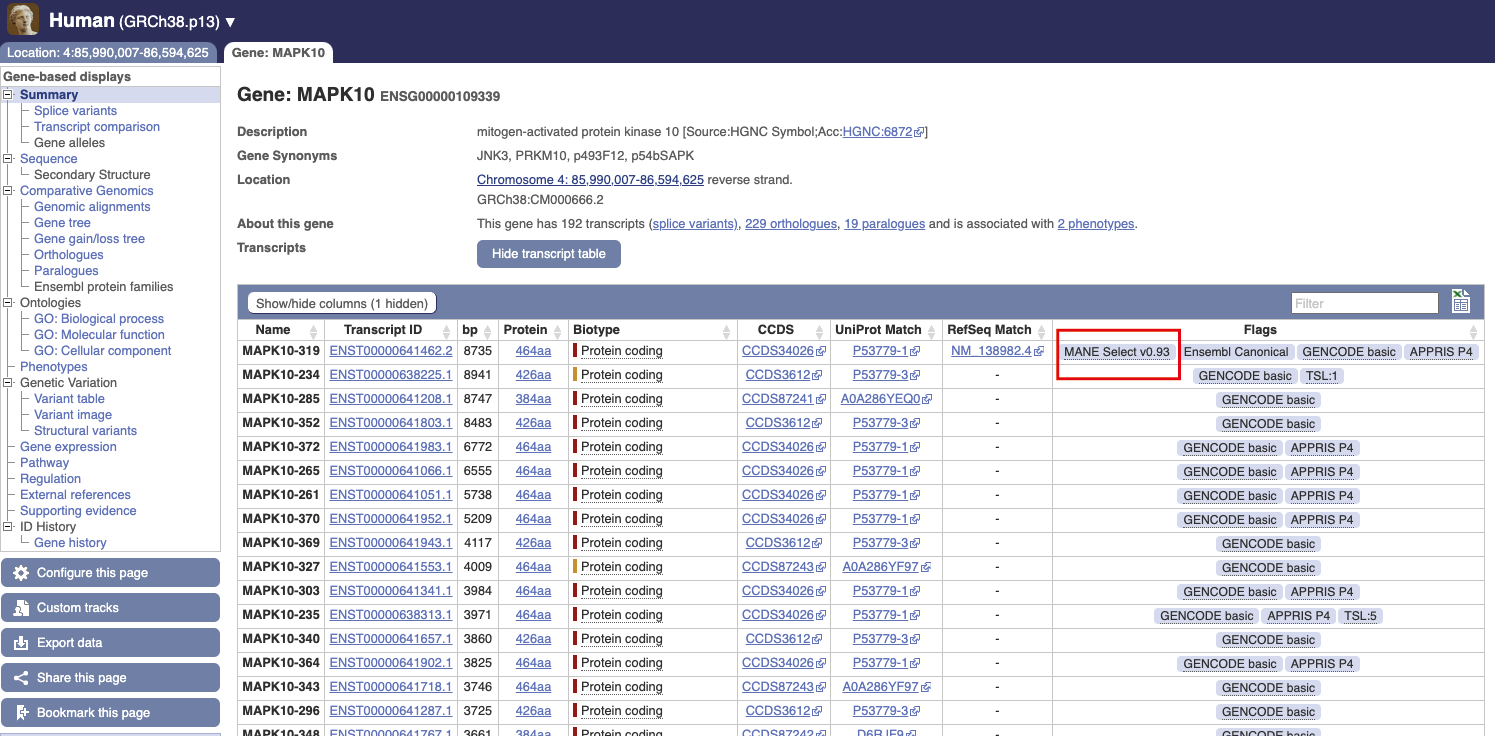
Many think that the gene annotation of the human genome was completed many years ago, and so we should know the number of protein-coding genes, but it’s not that simple. A lot of our time is taken with assessing and utilizing new datasets to make sure that we can add extra information to our geneset. For example, for well-known and well-characterised genes, such as MAPK10, we will add new alternatively spliced transcripts as new datasets become available. So, when we say that we are annotating genes what we are mainly doing now is adding new transcripts to already known genes. With new data constantly being created we will likely soon see hundreds of alternatively spliced transcripts per gene. There is always a bit of churn with the protein-coding gene numbers as we reassess the biotype of some genes in the light of new data, but we have a good idea that the protein-coding gene count is for human around 19,000 genes - but with an ever-expanding number of transcripts.
So, more data means more transcripts. But how do you know which transcript to prioritise? A recent project, the Matched Annotation from NCBI and EMBL-EBI (MANE) project, can help with this. As well as our Ensembl/GENCODE geneset, the NCBI’s RefSeq group also produce a high-quality human geneset. These two genesets are not identical due to many factors, including timing of release, methodology and interpretation of evidence. This can cause particular problems in a clinical context if the transcripts are assumed to be identical, for example for variant interpretation. The first product of the MANE project is the MANE Select transcript set, which identifies one representative transcript in the current reference human genome (GRCh38) for each protein-coding gene that is identical (5’ end, 3’ end, introns, exons) in both genesets. MANE transcripts have versioned Ensembl and RefSeq IDs that can be used interchangeably to provide a default display for browsers, resources and tools. We are currently working on a publication about this important project and hope to publish soon.
Further information can be found here: http://tark.ensembl.org/web/mane_project/