AlphaFold - an AI tool for protein structure prediction
HGNC ·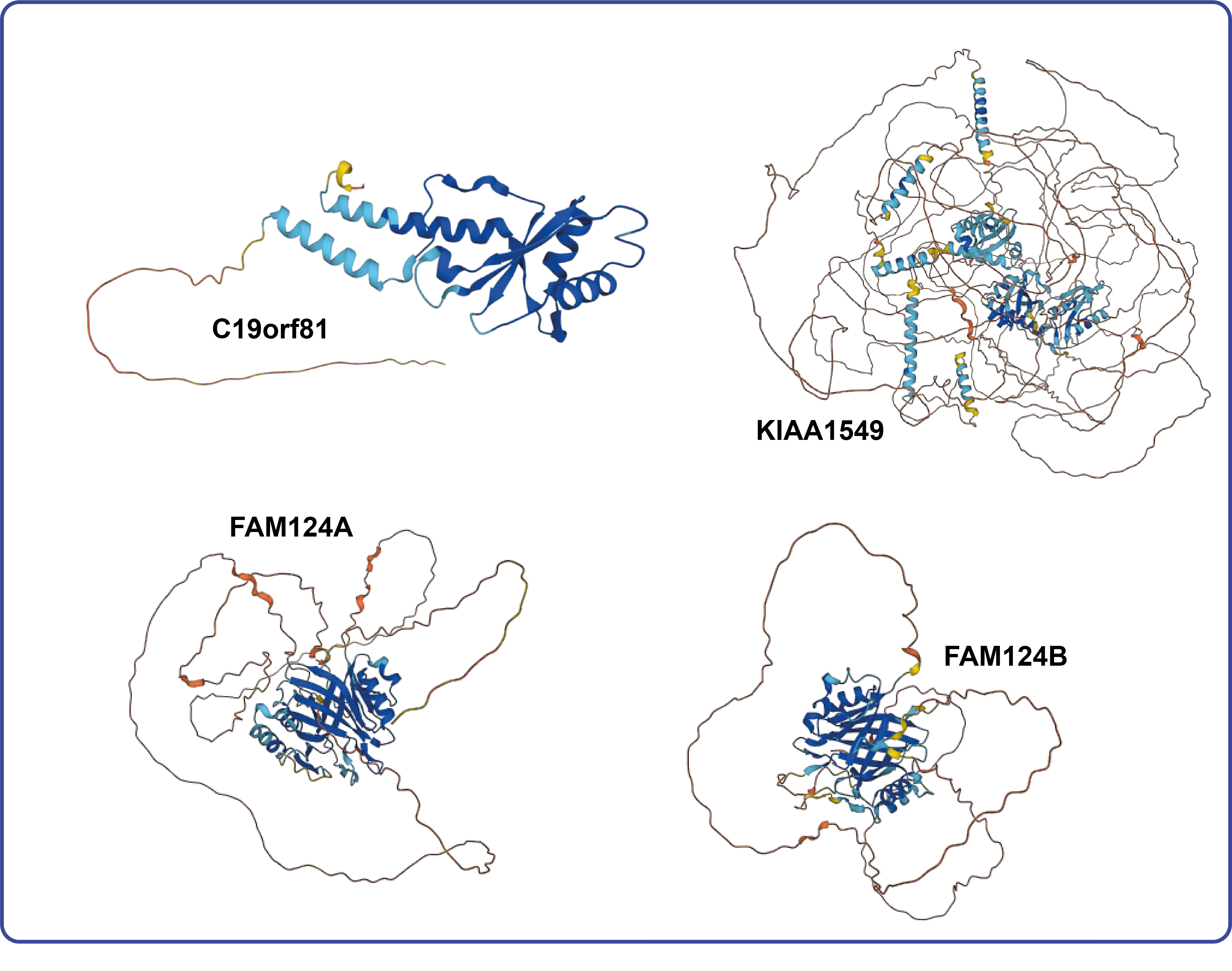
AlphaFold and Placeholder Symbols
Alphafold uses artificial intelligence to predict the 3D structure of proteins from their amino acid sequences. The system was developed as a collaboration between DeepMind and EMBL’s European Bioinformatics Institute (EMBL-EBI).
The HGNC are currently looking into using AlphaFold as a tool to help us in renaming genes that still have temporary “placeholder” symbols. HGNC policy has always been to view placeholder symbols as temporary assignments that should be updated when the function of a protein encoded by a gene is identified, and the majority of recent symbol changes have been updates of these placeholders (C#orfs, KIAAs and FAMs). You can find out more about placeholder symbols in one of our previous blog posts.
The HGNC recently worked with a group of researchers who used AlphaFold to help identify a set of genes that encode bridge-like lipid transfer proteins including two genes with placeholder symbols. The proteins encoded by this set of genes share structural similarity and can fold up to form a long rod-like structure with an internal hydrophobic groove. Some members of the group have been shown to transport lipids; for other group members this function is still putative, hence the term “family member” in some of the gene names. The group also found that trRosetta, another protein folding prediction program also produced similar results. The BLTP (bridge-like lipid transfer family) gene group can be seen here.
Introducing our Guest Post Author

David Nelson is a long term collaborator of the HGNC and a Professor at the University of Tennessee Health Science Center. He has been studying the evolutionary history of cytochrome P450s in species from across the tree of life for over 30 years, and you can read his previous guest post about the cytochrome P450s here.
In this guest blog post, David discusses a project that he is currently working on looking at the unannotated genes from the minimal cell chromosome, called JCVISyn3A/B from the J.Craig Venter Institute (JCVI). It is interesting to see that a high level of sequence similarity is not always a prerequisite for two proteins to share a very similar structure. Here’s David to tell us more:
Annotating the Mycoplasma minimal cell genes using AlphaFold and Foldseek Search
A minimal cell chromosome called JCVISyn3A and later JCVISyn3B was synthesized and booted up to make a free-living Mycoplasma cell.
In 2019 Breuer, et al. published “Essential metabolism for a minimal cell. eLife, 8, e36842. https://doi.org/10.7554/eLife.36842”. This paper described the JCVISyn3A chromosome as having 452 protein coding genes and a doubling time of 2 hours. A few non-essential genes were added for ease of genome manipulation and to help with robust growth to create the JCVISyn3B chromosome, which has 473 genes - 438 protein coding and 35 RNA coding genes.
At the time of publication 149 genes in the JCVISyn3A chromosome had no known function. The advent of Alphafold2 and a 3D structure search program named Foldseek Search has made it possible to search for 3D-structure matches that have annotation for family membership and/or functional information. The 149 sequences from the original publication were Blast searched against GenBank to get current accession numbers and any recent annotation. 96 of the 149 had some new annotation though some of it was generic such as lipoprotein or MFS transporter. That left 53 sequences with no annotation. We used the new AlphaFold database and Foldseek Search to find 3D homologs with annotation to fill in some of the lacking functional knowledge of the minimal cell.
The procedure required Blast search of UniProt for the closest hit, often 95% or better to the original sequence. UniProt was used because AlphaFold has predicted the structures of all of the proteins in UniProt (about 214 million). The UniProt accession number is the AlphaFold accession number. The UniProt accession was loaded into Foldseek Search and the Alphafold PDB file was uploaded and used to search the optional databases: AlphaFold/UniProt50 v3, AlphaFold/Swiss-Prot v2, AlphaFold/Proteome v2, PDB100 220722 and GMGCL 2204.
Hits were examined for reasonable 3D-alignments with good superposition of the query and subject over most of the length. These hits were checked for their annotations. It became clear that good 3D alignments with reasonable expect values did not have to have significant percent sequence identities. The sequence ID values may be in the 12-13% range for some 3D alignments.
Only about half of the 53 genes examined had new family or functional annotations. Many of the rest had strong matches to unannotated sequences, often from Mycoplasmas.
Here are some interesting examples of structural matches between sequences from the minimal mycoplasma proteome and proteins from other species.

The figure above shows a structural match between a mycoplasma protein from the minimal cell project and a S.pneumoniae protein named 'septation ring formation regulator EzrA'. This protein plays a role in bacterial cell division by negatively regulating FtsZ ring formation
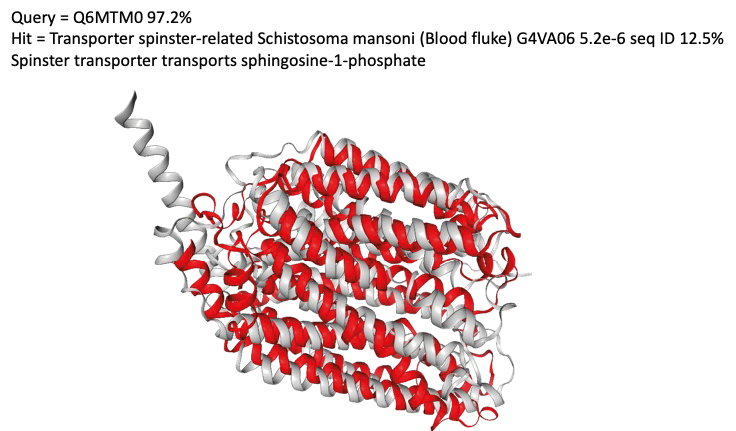
The figure above shows a structural match between a mycoplasma protein and a sphingolipid transporter from the parasitic blood fluke S.mansoni
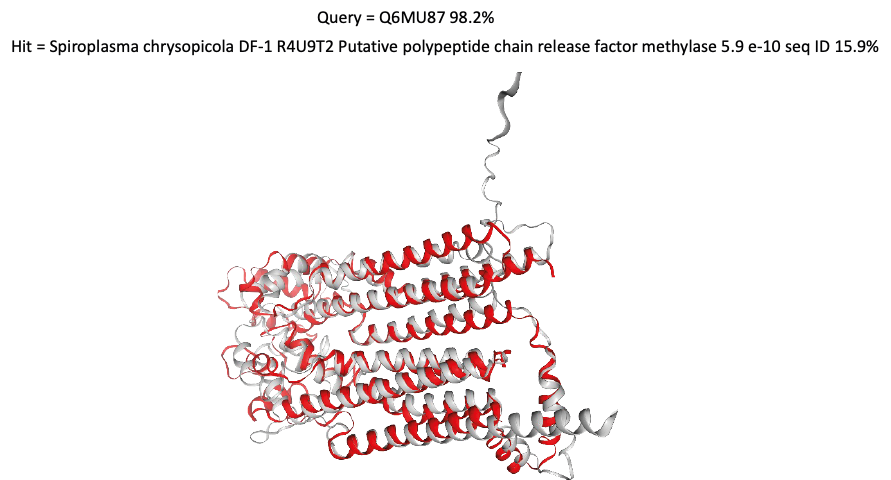
The figure above compares the structure of a mycoplasma protein with a protein from another Gram-positive firmicute bacteria called Spiroplasma chrysopicola. These proteins may act as release factors and be involved in the termination of the translation of synthesised proteins by the ribosomes, although this function is currently putative.
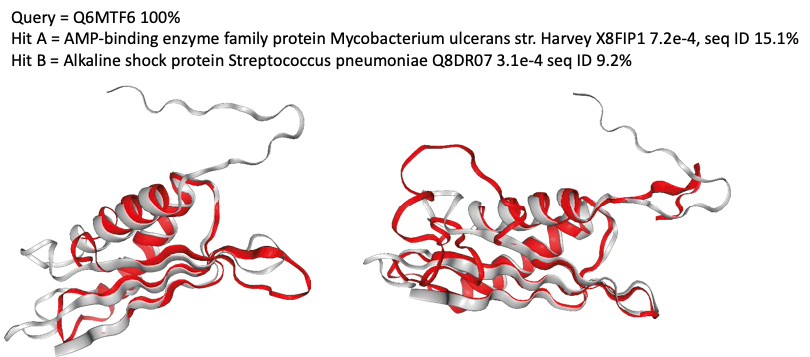
The figure above shows a structural match between a mycoplasma protein from the minimal proteome and two different proteins: an AMP-binding enzyme from M.ulcerans and an alkaline shock protein from S.pneumoniae.
If you would like to see the rest of David’s slides with more examples you can view them here.
Placeholder Symbols: we want your data!
We hope that you have enjoyed reading this blog post. If you have functional data about a gene that currently has a placeholder symbol, then please get in touch with us at hgnc@genenames.org so that we can work with you to assign appropriate new nomenclature.